
Alfonso D’Apuzzo – Napoli, Gjeorgjina Kuli Lito – Tirana, Doina Anca Plesca – Bucharest, Basim Alzoubi – Amman, Alketa Hoxha Qosia – Tirana, Maria Vendemmia – Napoli
Abstrat
The syndrome of adrenal insufficiency, alacrima, achalasia and neurologic deterioration (Allgrove Syndrome) was first described by Allgrove et al in 1978. It is an autosomal -recessive disorder. ADRACALIN (AAAS), (12q13 cromosome), is the altered gene enconding the protein ALADIN.responsibile for the clinical presentation. Neurologic involvement has been described in the form of a mixed pattern of upper and lower motor neuron dysfunction, impaired autonomic function, and some degree of intellectual deterioration..We describe a girl with complete syndrome (episode of severe hypoglicemia with seizures, hyperpigmentation, alacrima, progression of neurologic impairment, and peripheral poliyneuropathy, achalasia of cardia), in whom brain MRI revealed periventricular gray matter heterotopias. Molecolar genetic analysis showed mutations of the AAAS gene, that encodes the Nuclear pore complex protein ALADIN. Extensive biochemical and metabolic investigations have not revealed the cause of this disorder. Treatment: Hydrocortisone, Cortinef, Topical eye lubricants, rehabilitation for neurologic symptoms.
Case report
Una ragazza di 10 anni, aveva presentato, all’età di anni 2 e 6 mesi, episodi convulsivi ipoglicemici, aumento progressivo della pigmentazione cutanea dall’anno precedente, ed alacrimia sin dalla nascita, All’età di 3 anni e 6 mesi episodi di vomito con disfagia che determinava mancato aumento di peso. La Xgrafia con bario dell’apparato gastrointestinale e manometria esofagea avevano evidenziato marcata acalasia per cui veniva sottoposta alla miotomia di Heller con fundoduplicatio, con cessazione della sintomatologia. All’età di 9 anni iniziava a lamentarsi di dolori forti alle gambe e alle mani. L’esame clinico rivelava assottigliamento dei muscoli distali alle estremità superiori ed inferiori, debolezza muscolare generalizzata con precoce piede cavo a sinistra, ed aumento dei riflessi profondi e risposta plantare in estensione. Ipercheratosi palmare con aumento delle pieghe cutanee. I test funzionali autonomici evidenziavano diminuzione dell’attività parasimpatica. Non aveva ipotensione posturale, non disturbi della sudorazione ma ipotensione labile con crisi ipertensive. Gli studi di conduzione nervosa rivelavano conduzione prolungata, indicativa di demielinizzazione, l’elettromiogramma evidenziava atrofia muscolare neurogenica. EEG era normale.
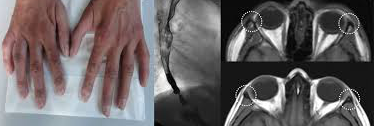
Era nata a termine da genitori consanguinei, gravidanza e parto erano stati normali, il peso alla nascita era di 2800 grammi. I genitori descrivevano ritardo dello sviluppo psicomotorio: posizione eretta a 18 mesi, primi passi a 24 mesi, uno spiccato ritardo del linguaggio (prime parole a 30 mesi.) IL peso e l’altezza erano al 10 ° perc. per l’età. La sua circonferenza cranica era -3 DS. L’esame fisico evidenziava una iperpigmentazione della faccia, collo, e arti. Gli esami endocrinologici rivelavano insufficienza adrenocorticale con test da stimolo all’ACTH, che non evidenziava elevazioni del cortisolo con livelli di meno di 15 nmol/L al tempo 0 e al 60° minuto. Bassi livelli di aldosterone con valori di 0,007nmol/L. Dopo trattamento con Idrocortisone 15mg/m2, la paziente presentava aumento dell’appetito, e scomparsa della iperpigmentazione. La risonanza nucleare magnetica encefalospinale evidenziava prominenza degli spazi cerebrospinali, solchi e noduli subependimali lungo i margini dei ventricoli laterali. I noduli erano isotensi con la sostanza grigia corticale in T1 e T2., patognomonici di eterotopia della sostanza grigia subependimale. Gli esami metabolici per il lattato, piruvato, ammoniemia, aminoacidemia, aminoaciduria, organicoaciduria, acidi grassi a catena molto lunga, l’acido fitanico, acido pipecolico, dosaggio ematico della vitamina B12 e dell’acido folico erano nella norma. Le indagini genetiche evidenziavano mutazione del gene ADRACALIN(AAAS), sul cromosoma 12q13, che codifica per la proteina ALADIN, responsabile della sindrome
Discussione
La sindrome è caratterizzata dai seguenti caratteristici sintomi: insufficienza adrenocorticale, acalasia, alacrimia. Tali sintomi variano nel tempo e sono accompagnati, più o meno, lentamente da progressivo deteriomento neurologico. Quest’ultimo include un misto pattern di disfunzione dei neuroni motori superiori ed inferiori con iperreflessia, aumento del tono muscolare, risposta plantare in estensione, muscoli marasmici, disturbo della funzione autonomica, con ipotensione posturale, ridotta sudoraziione, e riflessi cardaci abnormi. La nostra paziente ha la sindrome completa, con alacrimia presente sin dalla nascita, insufficienza adrenocorticale all’età di 2 anni, acalasia a 4 anni e progressivo danneggiamento dei neuroni motori superiori ed inferiori dopo i 9 anni.
LA SINDROME DI ALGROVE, va in diagnosi differenziale con:
- I disordini perossomiali (Adrenoleucodistrofia e adrenomieloleucodistrofia, in cui sono alterati gli acidi grassi a catena molto lunga, acido fitanico e pipecolico rispettivamente.
- Disordini autoimmuni con insufficienza surrenalica (sindrome APECED) per la assenza di autoanticorpi anti-corteccia surrenalica e a anti21-idrossilasi.
- Sindrome Antifosfolipidi per la asssenza degli anticorpi antifosfolipidi
- Sindrome adrenogenitale per i valori normali di 17 OH idrossilasi.
Bibliografia
Allgrove J, Clayden GS, Grant DB, McCauley JC: Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet 1978;1:1284-1286
Ambrosiano MM.. Geneiser NB, Bangaru BS et al: The syndrome of achalasia of the esophagus, ACTH insensitivity and alacrima. Pediatr Radiol 1986;16:328-329
Tsao CY, Romshe CA, Lo WD et al:Familial adrenal insufficiency, achalasia,, alacrima, peripheral neuropathy, microcephaly, normal plasma, very long chain fatty acids, and normal muscle mitochondrial respiratory chain enzymes. J. Child Neurol 1994:9:135-158




